
Генная терапия нервно-мышечных болезней: как лечебные гены возвращают людям подвижность
Спинальная мышечная атрофия, миодистрофия Дюшенна и болезнь Помпе — это тяжелые нервно-мышечные заболевания (НМЗ), обусловленные генетикой. Часто эти болезни начинаются в детстве и быстро приводят к потере способности передвигаться, инвалидности и смерти. На сегодняшний день самый прогрессивный метод их лечения — генная терапия, которая умеет «чинить» неисправные процессы на молекулярном уровне. Рассказываем, как современные геномные технологии продлевают жизнь людей с нейромоторными нарушениями.
18+
Редакция журнала «Нож» утверждает, что настоящая научно-исследовательская статья имеет исключительно культурную ценность, предназначена для использования в научных целях либо в образовательной деятельности и не является медицинской рекомендацией.
AAV-доставка лечебных генов
Нервно-мышечные болезни — обширная группа заболеваний, для которых характерна прогрессирующая мышечная слабость, нарушение моторных функций и двигательная инвалидизация. Прогресс в геномных исследованиях позволил выявить наследственные дефекты, отвечающие за развитие многих НМЗ, и разработать методы их лечения.
Существует несколько генотерапевтических стратегий лечения нервно-мышечных заболеваний, из которых ведущая — AAV-терапия, то есть доставка лечебного генетического материала к целевым клеткам с помощью рекомбинантного аденоассоциированного вируса. Эта методика уже более 20 лет тестируется в доклинических и клинических испытаниях.
Если в каком-то гене произошла «поломка», послужившая причиной заболевания, то болезнь можно вылечить или облегчить, если ввести в целевые клетки рабочую копию дефектного гена. Здесь на помощь приходят векторы — системы доставки, транспортирующие генетический материал к органам и тканям. Самые популярные системы доставки — вирусы, которые с легкостью проникают в клетки хозяина.
В генотерапии нервно-мышечных расстройств чаще всего используется аденоассоциированный вирус (AAV), у которого есть ряд преимуществ перед другими вирусами-доставщиками:
- Непатогенность. AAV не может самостоятельно вызывать инфекционные заболевания. Он становится опасным только при наличии вирусов-помощников — аденовируса, ВПГ, ВПЧ.
- Способность эффективно распространяться в различных тканях, включая нейроны и мышцы, проникать в делящиеся и неделящиеся клетки.
- Стабильность. AAV устойчив к воздействию тепла, кислому рН и протеазам.
При создании вектора AAV делают еще более безопасным: из него достают гены, отвечающие за репликацию и интеграцию в геном хозяина, и заменяют их на лечебные.
При нервно-мышечных расстройствах чаще всего применяется внутривенное введение: благодаря кровотоку препарат распространяется по всему организму и воздействует на группы мышц. Но в этом случае для достижения лечебного эффекта требуются высокие дозы препарата, что делает его токсичным. Кроме того, при внутривенном введении он достигает не только целевых мышц, но также проникает в печень, сердце и почки, что может вызывать осложнения.
Нервно-мышечные расстройства связаны с патологиями нервной системы, поэтому геннотерапевтический препарат должен уметь преодолевать ГЭБ — функциональный барьер, отделяющий ЦНС от кровеносной системы. Некоторые серотипы AAV умеют это делать, однако при внутривенном введении их воздействие на ЦНС низкое. Исследователи решают эту проблему несколькими способами:
- Введением препаратов в спинномозговую жидкость (ликвор), что позволяет одновременно воздействовать и на нервную систему, и на мышцы. В таком случае снижается дозировка, а значит, и токсичность препарата.
- Новыми серотипами AAV с повышенным тропизмом к ЦНС и мышцам. Ученые создают штаммы с новыми свойствами, влияющими на эффективность и безопасность лечения. Изменяя капсиды вируса, можно увеличить его тропизм к целевым клеткам или, наоборот, снизить воздействие на нецелевые органы.
На сегодняшний день большинство испытаний генной терапии сосредоточено на трех НМЗ: спинальная мышечная атрофия, миодистрофия Дюшенна, болезнь Помпе. Наибольшие успехи достигнуты в лечении первых двух заболеваний.
Перспективная терапия для СМАйликов
Спинальная мышечная атрофия (СМА) — группа редких заболеваний, характеризующаяся дегенерацией мотонейронов спинного мозга. СМА подразделяется на несколько типов, самый тяжелый — 1 тип. Дети с этой формой болезни не могут сидеть самостоятельно, большинство из них не доживают до двух лет. При других вариантах СМА люди живут гораздо дольше.
Виновник СМА был выявлен в 1995 году. Им оказался ген, который кодирует белок SMN, отвечающий за выживаемость мотонейронов. У людей имеются две практически одинаковые копии этого гена: SMN1 и SMN2.
У СМАйликов происходят мутации или потеря функции SMN1. Казалось бы, если SMN1 не работает, то его может «выручить» SMN2. Однако у последнего есть дефект, приводящий к синтезу укороченной и нестабильной формы SMN-белка.
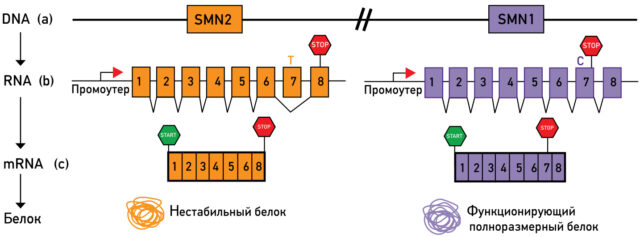
На сегодняшний день разработаны две стратегии генотерапии СМА:
- Устранение дефекта SMN2.
- Доставка к целевым клеткам нового функционального гена SMN1.
В 2016 году FDA был одобрен Нусинерсен (торговое название Spinraza) — первый генотерапевтический препарат для лечения СМА. Это антисмысловой олигонуклеотид, вводимый в спинномозговую жидкость и устраняющий дефект SMN2.
Клиническое исследование препарата с участием 122 детей в возрасте до 7 месяцев со СМА 1 типа показало улучшение двигательной активности у 41% испытуемых. Кроме того, генная терапия улучшала выживаемость пациентов.
Другие исследования продемонстрировали эффективность Spinraza в терапии более легких форм СМА. В России Spinraza одобрили в 2019 году.
У детей со СМА 1 типа симптомы заболевания при рождении отсутствуют, задержка развития двигательных функций возникает в течение первого полугода. При других формах симптоматика стартует еще позже. Исследование NURTURE показало, что терапия Spinraza на доклиническом этапе эффективнее, чем уже после появления симптомов.
В большинстве стран, включая Россию, возможно пресимптоматическое лечение СМА, что оправдывает проведение скрининга новорожденных на наличие болезни. Такой скрининг введен в 46 штатах США и многих европейских странах. В 2023 году он был запущен и в России.
После Spinraza были одобрены еще два генотерапевтических препарата для лечения СМА. Первый, Risdiplan, работает так же, но вводится пероральным способом. Второй — Zolgensma, доставляющий в организм полноценный ген SMN1, упакованный в AAV-вектор. Препарат вводится в вену всего один раз.
В 1,5-годовом исследовании оценивалась безопасность и эффективность препарата. В экспериментальную группу вошли 22 ребенка со СМА 1 типа, средний возраст составил 3,7 месяца. Результаты были следующими:
- 91% детей выжили и не нуждались в постоянной вентиляции легких. При естественной истории заболевания только четверть детей в возрасте 14 месяцев остаются в живых без поддерживающей дыхательной терапии.
- У 95% детей показатель CHOP INTEND превышал 40 баллов. Без лечения дети со СМА в возрасте 6 месяцев и старше почти никогда не достигают таких показателей.
- Более половины смогли сидеть без поддержки не менее 30 секунд. СМАйлики, которые не получают лечения, не могут сидеть самостоятельно.
В 2022 году Zolgensma поступила в оборот в России, стоимость одной дозы составила более 82 млн рублей. Лечение получили более 150 детей, чьи истории демонстрируют, что препарат положительно влияет на двигательную функцию, качество и продолжительность жизни.
Некоторые СМАйлики, дожившие до взрослого возраста, готовы пойти на всё, чтобы избавиться от болезни. Один даже хотел пересадить свою голову на здоровое донорское тело. Трансплантацию должен был провести итальянский хирург Серджио Канаверо, однако операция до сих пор не состоялась.
Надежда для людей с миодистрофией Дюшенна
Миодистрофия Дюшенна (МДД) — тяжелая наследственная форма миодистрофии, отличающаяся ранней манифестацией и быстрым развитием. Часто первые симптомы появляются в 3–5 лет, к 12 годам многие дети теряют подвижность. Большинство не доживают до 20–30 лет, смерть наступает в результате сердечной и дыхательной недостаточности.
Эффективных лекарств от болезни нет. Многие лечебные стратегии тестируются, но AAV-терапия демонстрирует наиболее многообещающие результаты.
Причина заболевания — множественные «поломки» гена DMD, которые приводят к дефициту белка дистрофина, играющего важную роль в функционировании мышц. Так как при заболевании ген поврежден многократно, устранить проблему можно внедрением в клетки здорового гена, кодирующего дистрофин. Однако существует препятствие: DMD — крупнейший человеческий ген, который невозможно «впихнуть» в вирусный вектор.
Частично решить проблему удалось с помощью микродистрофиновой терапии. У МДД есть более легкая форма — мышечная дистрофия Беккера, которой тоже свойственны слабость мышц и «поломка» гена. Однако при ней он поврежден гораздо меньше. Известен случай, когда пациент с мышечной дистрофией Беккера дожил до преклонного возраста, хотя ген DMD у него был уменьшен наполовину. Этот случай подтолкнул ученых к созданию терапевтического гена микродистрофина. Микродистрфиновая терапия не избавляет от МДД, но облегчает болезнь и замедляет ее прогрессирование.
В настоящее время тестируются три препарата с микродистрофином, в частности, SRP-9001, прошедший вторую фазу испытаний.
В экспериментальную группу вошли дети с МДД в возрасте от 4 до 8 лет. Стабилизация двигательной функции сохранялась у них в течение двух лет после окончания лечения.
Продолжаются дальнейшие исследования по оценке безопасности и эффективности препарата в более широких популяциях.
Еще один перспективный метод генной терапии МДД — пропуск экзонов, участков ДНК с набором инструкций для синтеза белка. Мутации и делеции, наблюдаемые при миодистрофии Дюшенна, могут затрагивать один или несколько экзонов, что приводит к выработке нефункциональной формы дистрофина. Пропуск дефектных экзонов позволяет восстановить нормальную работу остального гена и запустить синтез укороченной, но более функциональной формы белка.
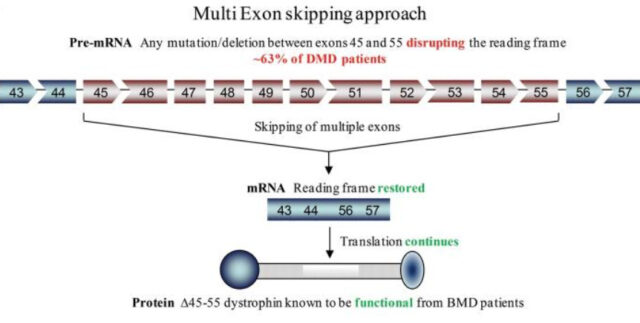
Пропуск экзонов достигается введением в организм антисмысловых олигонуклеотидов, «маскирующих» необходимые участки. Один из препаратов с подобным действием одобрен FDA. Клинические испытания демонстрируют его эффективность: на 48-й неделе лечения у пациентов увеличивается подвижность мышц. В группе плацебо, наоборот, отмечалась отрицательная динамика.
В доклинических испытаниях с мышами для «вырезания» экзонов используется система редактирования генома CRISPR / Cas9, которую называют генетическими ножницами. После редактирования дефектных генов у грызунов частично восстанавливался синтез дистрофина в скелетных и сердечных мышцах и улучшилась подвижность скелетных мышц.
AAV-терапия болезни Помпе
Болезнь Помпе — смертельное наследственное заболевание, при котором из-за дефицита фермента кислой мальтазы накапливается нерасщепленный гликоген, что ведет к поражению спинного и головного мозга, повреждению гладких, скелетных и сердечных мышц.
Причина заболевания — мутации в гене GAA, который кодирует фермент кислой мальтазы. Ученые пытаются устранить этот дефект с помощью AAV-терапии, направленной на доставку функционального гена к целевым клеткам.
В настоящее время ведется как минимум 5 клинических испытаний AAV-терапии болезни Помпе. Американские медики тестируют препарат ACT-101В, доставляющий ген в гепатоциты. Исследование продемонстрировало безопасность и биологическую активность препарата — уровень фермента кислой мальтазы в мышцах у испытуемых увеличился более чем в два раза. К сожалению, лечение не улучшило двигательную функцию. Однако пока что тестировались низкие дозы препарата, дальнейшие исследования будут направлены на оценку эффективности более высоких дозировок.