
Съесть чужой мозг и превратить собственный мозг в губку: история открытия и изучения прионов — загадочных белков, которые иногда становятся убийцами
В Папуа — Новой Гвинее множество различных племен, но сегодня нас будет интересовать только племя форе: именно в его языке впервые появилось слово «куру», два основных значения которого — «порча» и «дрожь». Люди верили, что шаман из недружелюбного соседнего племени может наслать на них порчу, и тот, кому не повезло, считался обреченным: сначала он начинал мелко дрожать, мышцы сводило судорогой, и постепенно тело полностью переставало слушаться. Смерть наступала через несколько месяцев, максимум год, причем чаще всего куру поражала детей и женщин. Зловредные шаманы так и продолжали бы наводить ужас на соседей, если бы в 1957 году в племя форе не приехали два врача — представитель новозеландского министерства здравоохранения Винсент Зигас и американский вирусолог Дэниел Гайдузек. О том, что они обнаружили, рассказывает Зоя Андреева.
История открытия прионов
Дэниел Гайдузек, хоть и был молодым ученым, уже успел на тот момент зарекомендовать себя — окончил Гарвард, а в Калифорнийском университете даже работал с нобелевским лауреатом Лайнусом Полингом. После такого старта карьеры Гайдузеку были открыты все пути, и несколько лет он работал в Гарварде, после чего уволился и перебрался сначала в Австралию, а затем в Новую Зеландию. Там он познакомился с Винсентом Зигасом, который и рассказал вирусологу о непонятном заболевании, считающемся местными порчей. Заинтересовавшись и выучив основы языка, Гайдузек вместе с Зигасом отправились в племя форе, где прожили больше года, изучая загадочное проклятие.
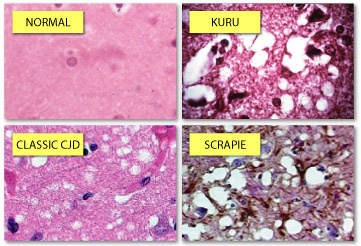
Изучая куру, ученые вскрыли огромное количество трупов и довольно быстро пришли к выводу, что у всех погибших наблюдалась одна и та же картина: их мозг буквально превращался в губку. Так под подозрение попал именно мозг, а наблюдение за поведением племени помогло разгадать, как болезнь распространялась. Оказалось, что в племени активно практиковался каннибализм, причем в основном именно мозга: считалось, что поедание мозга — это своего рода дань уважения к усопшему и что именно так можно «впитать» его хорошие качества, став более сильным, храбрым или умным. Чаще всего мозг «доставался» женщинам и детям, поэтому среди них заболеваемость была самой высокой.
Банальный эксперимент подтвердил догадку: прекратив массовый каннибализм в одном отдельно взятом племени, Гайдузек и Зигас снизили заболеваемость до нуля.
Гайдузек довольно быстро заметил инфекционный характер заболевания и даже проводил эксперименты по осознанному заражению куру-подобным состоянием, используя в качестве подопытных шимпанзе.
Как передавалась куру, удалось выяснить быстро, но патоген, отвечающий за развитие заболевания, долго оставался неизвестным. Сам Гайдузек считал, что губчатая энцефалопатия — разрушение мозга, когда он становится похожим на губку, — вызывается вирусом из семейства лентивирусов, или так называемых медленных вирусов (одним из представителей этого семейства является вирус иммунодефицита человека). Именно о медленных вирусах Гайдузек рассказывал во время своей нобелевской лекции — в 1976 году он удостоился премии по физиологии и медицине, разделив ее с Барухом Бламбергом, описавшим вирус гепатита В (любопытно, что Винсенту Зигасу премия не досталась).
Но были и противники вирусной теории. Во-первых, неизвестный патоген, выделенный из мозга, продолжал заражать подопытных животных даже после длительного нагрева и после обработки. Всех исследователей удивляло, что способность к заражению у этой субстанции, выделенной из мозга больных животных, сохранялась и после сильного длительного нагрева, и после обработки детергентами и окислителями. Не действовало на патоген ни УФ-излучение, ни другие обычные способы деактивации вирусов. Это наталкивало на мысль, что либо этот «вирус» не содержит нуклеиновой кислоты — иначе она распадалась бы под столь жестким воздействием, либо патоген вовсе не является вирусом. Кроме того, на патоген не действовала и радиация. Известно, что доза радиации, необходимая для уничтожения молекулы, зависит от ее размера: чем она меньше, тем меньше вероятность попадания в нее заряженной частицы. Так было показано, что молекула, вызывающая куру, меньше, чем вирус.
В итоге в 1967 году сразу два британца — Джон Гриффит и Тивах Альпер — независимо друг от друга выдвинули белковую теорию развития заболевания. Патоген, считали они, не вирус и не какая-либо другая известная форма жизни, а «неправильный» белок.
Эти статьи прочитал еще один герой нашей истории — американский врач Стэнли Прузинер, который впоследствии и получил Нобелевскую премию за открытие прионов и описание механизма заражения.
Стэнли Прузинер тоже успел поработать в нескольких крупных госпиталях и университетах, а в 1972 году, когда он практиковал в отделении неврологии Университета Калифорнии в Сан-Франциско, у него умерла пациентка. Вскрытие показало, что умерла она от всё той же губчатой энцефалопатии, но не от куру, а родственного заболевания. Изучение литературы показало, что это заболевание — болезнь Крейтцфельдта — Якоба — было описано еще в 1920-х годах и по развитию и клинической картине очень напоминало куру — точно так же, как и при куру, болезнь развивалась достаточно медленно, но неотвратимо, поражая нервную систему и мозг. Прузинер понял, что за прошедшие пятьдесят лет патоген так и не был найден, и поставил себе амбициозную задачу раскрыть эту тайну.
По всей видимости, умения работать и видеть перед собой цель Прузинеру было не занимать, и через 10 лет, в 1982 году, он опубликовал статью, в которой прямо указывал в качестве патогена «неправильный» белок. Он назвал его прионом, от латинских слов proteinaceous и infectious, что в переводе обозначало просто «инфекционный белок» (proteinacious infectious particle).
Эксперименты шли долго, и начал Прузинер с того, что попытался выяснить, к чему патоген неустойчив, — уже было известно, что он не реагирует ни на известные детергенты, ни на нагревание, ни на УФ-излучение. И ответ пришел: оказалось, что патоген чувствителен к ионизирующему излучению, но только в присутствии кислорода. Такое поведение наблюдалось для большинства гидрофобных белков, и это косвенно подтвердило идею Гриффита и Альпера.
Прузинер пошел дальше и в конце концов сумел выделить из пораженного мозга патоген — им действительно оказался белок. В классификации его назвали PrP, а чуть позже методом геномного секвенирования на 20-й хромосоме нашли и ген, отвечающий за его экспрессию. Это открытие было достаточно неожиданным: оказалось, что прион в норме присутствует в нашем организме. Более того, его аналоги обнаружились и у других видов, причем не только у млекопитающих, но и у рыб — то есть белок еще и достаточно древний!
И тут случилось событие, подстегнувшее научное сообщество к еще более тщательному исследованию прионов. В 1986 году в Великобритании зарегистрировали губкообразную энцефалопатию крупного рогатого скота (бешенство коров). Эпидемия продолжалась больше десяти лет, и на ее пике в 1992 году заболеваемость составляла 1000 случаев в неделю. К 1997 году болезнь поразила уже миллион животных, причем заболевание стало проявляться и у людей, употреблявших зараженную говядину в пищу. В конце концов, после жестких реформ в сфере сельского хозяйства, скрепи — так назвали заболевание — удалось купировать, и в нулевых заболеваемость упала практически до нуля. Но интерес к прионам остался.
Зачем они нужны?
Если гены, кодирующие прионы, есть в организме, то получается, что есть и «здоровые» прионные белки. Такой «нормальный» белок назвали PrPC. Сразу же встал вопрос: зачем он нужен в норме и как именно происходит заражение? Прузинер показал, что для развития прионного заболевания вовсе не обязательно наличие патогена, пришедшего в организм извне, — бывают и заболевания, которые возникают при мутации в гене PRNP, — это наследственные прионные заболевания. Кроме того, изменения могут возникнуть и сами по себе — спорадически, то есть спонтанно, причем механизм этой «внезапности» до сих пор не ясен, а вариантов мутаций много.
На наследственный способ заражения приходится 13% случаев, на приобретенный — менее 1%; все остальные — 87% — относятся именно к спорадическому способу заражения.
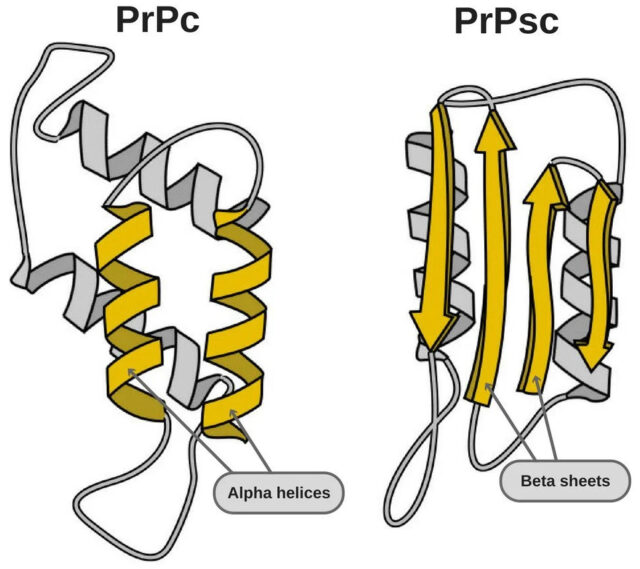
Важным шагом стало и открытие структуры приона и его «нормального» родственника. Первичная структура — линейная последовательность аминокислот — у обоих белков одинаковая. А вот вторичная и третичная структуры, образующиеся при сворачивании линейной цепи в клубок, — отличаются. Отличия нашлись в структурах под названием спирали: у нормального приона вторичная структура содержит 42% α-спиралей и почти не содержит β-складок (на них приходится не больше 3%), в то время как у дефектного белка PrPSc (он появляется у крупного рогатого скота при скрепи) наблюдается совсем другое соотношение — 30% α-спиралей и целых 43% β-складок! Именно это и позволило выдвинуть предположение, что «проблемность» приона зависит от неправильного фолдинга (сворачивания) изначально вполне правильного белка. Эта гипотеза со временем стала основной, хотя первооткрыватель куру — Дэниел Гайдузек — до конца жизни придерживался теории медленных вирусов. Что ж, в конце концов, он был вирусологом.
Попадая в организм, мутантный прион связывается с нормальным прионом и изменяет его конформацию. Вероятно, патогенный прион имеет большее сродство с обычным прионом и прикрепляется к нему. При этом происходит принудительное механическое изменение конформации «нормального» приона, иначе полная связка не была бы возможна. Такой олигомер далее присоединяет всё больше и больше «нормальных» прионов. При этом часто наблюдаются отложения амилоидных бляшек, таких же, как, например, при болезни Альцгеймера (именно отложение амилоидов в клетках мозга считается одним из основных провоцирующих болезнь Альцгеймера факторов).
И тут новый сюрприз — функция «нормального» приона до сих пор точно не ясна, хотя есть множество предположений и возможных сфер его деятельности. Зачем он нужен и почему кодируется в нашем собственном геноме, неся при этом потенциальную опасность? Попытки выяснить функциональность, конечно, были. Нокаутные мыши (то есть мыши, у которых выключен определенный ген) были вполне жизнеспособны. Потом попытались выяснить, влияет ли на заболеваемость наличие или отсутствие собственного прионного белка. Чтобы это проверить, нокаутным мышам вводили гомогенат мозга мышей, больных аналогом куру у животных. И мыши не заболевали, то есть отсутствие «собственного» спасало от заражения.
Последние эксперименты показали, что «нормальный» прион синтезируется в эндоплазматической сети. Потом белок транспортируется на поверхность клетки и закрепляется на ней. Структурный анализ также показал, что он может связывать ионы двухвалентных металлов — меди, цинка, никеля, марганца.
Согласно основной теории, «нормальный» прион может выполнять сигнальные функции, препятствовать окислительному стрессу, принимать участие в межклеточном узнавании и клеточной активации и даже участвовать в формировании памяти.
Скорее всего, в процессе спонтанной мутации приона клеточная мембрана тоже играет роль. По крайней мере, удаление холестерина из скопления прионов ингибирует переход «нормального» белка в патогенную форму.
Итак, ученые смогли выяснить, что из себя представляет прион, откуда он может взяться в здоровом организме, и даже более-менее определили его исходное местонахождение. Но вот триггер превращения пока что не найден.
Заключение
Пока что человечество не умеет лечить прионные заболевания. Если вспомнить, что еще в 2011 году была показана возможность воздушно-капельной передачи прионного заболевания, то возникает вполне резонный вопрос: можно ли ожидать среди людей такую же эпидемию, как среди британских коров? На самом деле вряд ли.
Распространенность прионных заболеваний, особенно спорадически возникших, довольно мала. Употребление в пищу мозга поверженного врага тоже дело достаточно редкое, а мясо в большинстве блюд хорошо обрабатывается. Остается лишь один неприятный вариант — использование прионов в качестве биологического оружия, но и здесь вероятность невысока: прионные заболевания имеют тенденцию развиваться медленно, иногда «выжидая» внутри организма много лет. Скорее всего, эпидемия нам не грозит.